VASP计算tips(备忘)¶
计算过程实时监测迭代情况:执行conv.sh脚本,然后用origin作图
计算过程中查看硬盘使用情况:df -h
Xshell上传文件:rz -E 下载文件sz filename
计算过程中查看CPU以及内存占用情况:top 1 2 退出top q
通常Windows编写的脚本上传到服务器上均会有问题,要进行格式转换才可以正常运行,
方法:Linux下安装dos2unix:
sudo apt install dos2unix运行命令进行转换:
dos2unix sub.sh转化为Linux格式
Linux下作图软件gnuplot非常常用,安装命令:
sudo apt install gnuplot查看VASP运行任务:
(base)host@ubuntu:~$ ps -ef |grep vaspVASP计算建议:
You have a (more or less) ‘large supercell’ and for larger cells it might be more efficient to use real-space projection operators. Therefore, try LREAL= Auto in the INCAR file. Mind: For very accurate calculation, you might also keep the reciprocal projection scheme (i.e. LREAL=.FALSE.).
对于较大的晶胞,使用实空间投影算子可能会更有效。因此,请在INCAR文件中尝试LREAL = Auto。注意:要进行非常准确的计算,您可能还需要使用倒数投影方案(即LREAL = .FALSE.)。
The determination of the symmetry of your systems shows a strong dependence on the tolerance parameter SYMPREC. This is often the result of “noise” in the structural parameters (lattice vectors and/or ionic positions). Set NWRITE=3 for more details.
确定系统的对称性强烈依赖于公差参数SYMPREC。这通常是结构参数(晶格矢量和/或离子位置)“杂音”的结果。设置NWRITE = 3以获得更多详细信息。
STOPCAR
目的是使正在运行中的VSAP程序主动停止计算,输出相应的文件后退出。STOPCAR非常简单。
如果它包含“LSTOP=.TRUE.”,则VASP在下一次离子循环时停止;
如果它包含“LABORT=.TRUE”,则VASP在下一次电子自洽时停止。
LSTOP=.TRUE.
LABORT=.TRUE.
停止之后可能会输出电荷密度、波函数等文件,但是在第二种情况下,电荷密度和波函数中可能并不是收敛的数值。
这个文件的作用与直接杀死程序是不同的,后者是强制停止计算,VASP不能及时输出相应的文件。
VASP计算遇到新旧电荷密度不同解决方法:
错误提示:BRMIX: very serious problems the old and the new charge density differ 解决方法:在vasp.pbs提交任务的脚本中加入 export I_MPI_ADJUST_REDUCE=3
计算能带工具使用方法:
#vaspktool# alias gk="~/vaspktool/gk.x" alias pb="~/software/ktool/pbnf.x" alias f="grep E-fermi OUTCAR" alias lv="grep -A3 'lattice vectors' OUTCAR"
VASP计算错误提示:ERROR: k-point generating vectors and reciprocal lattice are incommensurate. 解决方案:可设置ISYM=0(取消对称性),这个能是vasp6.1的一个小bug。
计算光学性质(optic)的时候报错:ERROR FEXCP: supplied Exchange-correletion table is too small, maximal index : 2902072。这个错误是关于交换关联函数表的大小不足。这通常与ENCUT值过大有关。
解决方案:
降低ENCUT值:
ENCUT = 500 改为较小值,比如: ENCUT = 400 或 ENCUT = 450
如果必须使用高ENCUT,可以增加ENAUG:
添加: ENAUG = 2000 # 增加辅助基矢的截断能
其他可以尝试的参数:
PREC = Accurate # 改为高精度模式 或 PREC = Normal # 保持正常精度
检查POTCAR:
确保POTCAR文件正确
确保POTCAR中的元素顺序与POSCAR一致
可以尝试使用不同版本的赝势
基本解决步骤:
首先尝试降低ENCUT值
如果问题仍然存在,添加ENAUG参数
如果还有问题,检查POTCAR文件 注意:
ENCUT值不要超过POTCAR中建议的最大值
过高的ENCUT会显著增加计算成本,但不一定提高精度
建议先用较小的ENCUT测试,再逐步增加直到收敛
问题:You are using a small complex shift. This can yield unreliable results when the Kramers-Kronig relation is used for systems with spin-orbit interactions. Please increase CSHIFT in the INCAR file.
这是VASP关于小复位移(small complex shift)的警告提示,我来翻译解释:
你正在使用一个很小的复位移值。在带有自旋轨道相互作用的体系中, 当使用Kramers-Kronig关系时,这可能会导致不可靠的结果。 请在INCAR文件中增加CSHIFT的值。
关于这个警告:
CSHIFT是什么:
是计算中使用的复位移参数
用于避免数值计算中的奇点问题
问题出现的情况:
当体系包含自旋轨道耦合(SOC)时
使用了Kramers-Kronig关系进行计算
CSHIFT值设置得太小
解决方案:
在INCAR文件中增大CSHIFT的值
通常建议从0.1开始尝试
注意事项:
这个问题主要影响带有自旋轨道相互作用的计算
如果没有包含SOC,这个警告可以忽略
如果包含SOC,建议认真对待这个警告
vasp中DFT+U设置
VASP计算中,采用LDA+U模型
U值由以下三个参数控制, LDAUL, LDAUU, LDAUJ LDAUL——对具体的p-/d-/f-轨道加U LDAUU——电子库伦相互作用项( on-site Coulomb interaction) LDAUJ——电子交换相互作用项( on-site exchange interaction) (U-J——有效U值)
具体参数的定义如下(假如体系中有两类原子La 和S,S不加U,La的f轨道修正(U-J)):
LDAU=T (控制计算中是否考虑在位库伦校正相) LDAUTYPE= 2 (+U的类型, 1|2|4; 2-默认值; U-J具有实际物理意义) LDAUL= -1 3 (控制具体的原子轨道上加U; -1, 不加U; 1-p轨道; 2-d轨道; 3-f轨道) LDAUU= 0 5.5 (U值由LDAUU-LDAUJ确定) LDAUJ= 0 0.5 LMAXMIX= 6 (对+U体系, 设置线性混合参数, f轨道设为6)
VASP加电场
参数: LDIPOL=.TRUE. IDIPOL=1/2/3(1=x,2=y,3=z) EFIELD= ×× (units V/A) IDIPOL也可以等于4 For IDIPOL=1-3, the dipole moment will be calculated only parallel to the direction of the first, seccond or third lattice vetor, respectively. The corrections for the total energy are calculated as the energy difference between a monopole/dipole and quadrupole in the current supercell and the same dipole placed in a super cell with the corresponding lattice vector approaching infinity. This flag should be used for slab calculations. For IDIPOL=4 the full dipole moment in all directions will be calculated, and the corrections to the total energy are calculated as the energy difference between a monopole/dipole/quadrupole in the current supercell and the same monopole/dipole/quadrupole placed in a vacuum, use this flag for calculations for isolated molecules. 更准确的说法是vasp官方指南的单位是F=qE 所以对应电场的单位是E=F/q 因而1eV/A对应的场强1V/A 一开始电场不要加的太大,电场的强度逐渐加上去。比如一开始做一个电场强度很小的计算,得到了收敛的电荷密度和波函数之后,再增加电场强度,读入上一步的电荷和波函数进行计算。。。。。这样的方式逐渐增加电场强度。 另外也要注意,这里所加的电场所能处理的体系,即电场的方向上是要有真空层的。 ISTART = 1 ICHARG = 1
VASP中(其他大部分DFT计算)加入电荷都是作为背景电荷加入,至于电荷如何再分布,不是依靠输入文件来指定的,而是通过波函数的优化实现的。所以不存在你希望加入一个电荷到某个特定的原子上去。这样的结果就是,如果这个加入的电子分布到你期望的原子上更稳定,那大部分会分配到这个原子周围。从经典角度,我们习惯用整数电荷以及电荷得失来分析问题,但从量化角度,电荷分布需要遵循能量最低化,需要全局化考虑,大部分情形下也不再是整数的。
vasp计算中用到的三种赝势:模守恒赝势,超软赝势,PAW赝势(按产生顺序)。
按方法不同分为USPP(ultrasoft pesudopotential,超软赝势)和PAW(projector augmented wave,投影缀加平面波),两种方法都可以相当程度地减少过渡金属或第一行元素的每个原子所必需的平面波数量。 按交换关联函数不同分为LDA(local density approximation,局域密度近似)和GGA(generalized-gradient approximation,广义梯度近似),GGA又分为PW91(Perdew -Wang 91)和PBE(Perdew-Burke-Ernzerhof)。 求解K-S方程时电子之间的交换关联泛函取局域密度泛函LDA(Rc处电子结构当成是密度相同的均匀电子气体来算)。 广义梯度泛函GGA(考虑了密度的梯度变化),但泛函的不同也会引起贋势的不同。 paw文件夹:PAW-LDA paw_gga文件夹:PAW-GGA-PW91 paw_pbe文件夹:PAW-GGA-PBE pot文件夹:USPP-LDA pot_GGA文件夹:USPP-GGA 选择某个目录进去,我们还会发现对应每种元素往往还会有多种赝势存在。这是因为根据ENMAX的大小还可以分为 Ga,Ga_s(soft),Ga_h,或者根据处理半芯态的不同还可以分为Ga,Ga_sv(s电子作为半芯态),Ga_pv的不同。 对于化合物(不同原子半径的元素混合)来说,PAW赝势比超软赝势精确度高。 US型赝势所需截至能较小,计算速度快,PAW赝势截至能通常较大,而且考虑的电子数多,计算慢,但精确度高。 在做具体的计算之前,针对需要计算的性质选取不同的贋势做下测试。对于不同贋势得到的结果,关注其最终的能量差,而不是绝对能量。
VASP结构优化过程中查看能量和力收敛
两个shell脚本,是用来查看vasp结构优化过程中能量和力等收敛情况的。
第一个脚本比较简单:
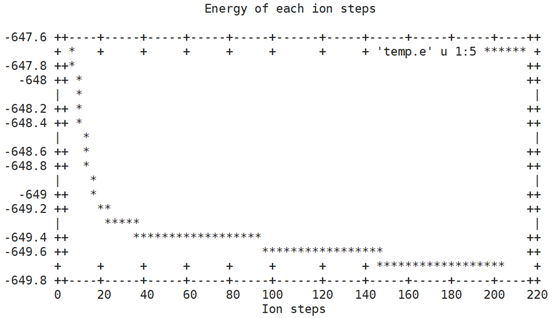
#!/bin/sh #edit by lipai@USTC #plot energies of structure in optimization awk '/E0/{if ( i<=5 ) i++;else print $0 }' OSZICAR >temp.e gnuplot <<EOF set term dumb set title 'Energy of each ion steps' set xlabel 'Ion steps' set ylabel 'Energy(eV)' plot 'temp.e' u 1:5 w l EOF #rm temp.e
其过程就是查看OSZICAR文件中每一离子步的能量,然后用gnuplot把数据画出来。
效果如下:
第二个脚本画两个图:
一个图是计算每一步的结构离初始的POSCAR结构有多远(需要用到vtst的脚本dist.pl,输入which dist.pl确定此脚本在系统环境路径里)第二个图是画出每一步中原子所受最大力。第二个图中的力对应于INCAR中的EDIFFG参数,某个离子步中的最大力小于EDIFFG的绝对值,就达到收敛结束计算。
效果如下:
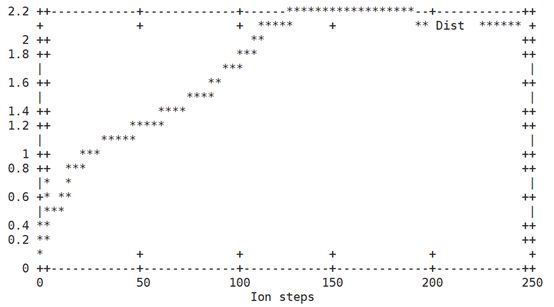 相对于初始结构RMSD
相对于初始结构RMSD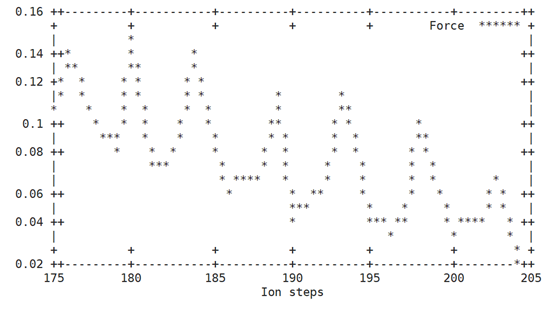 最大原子受力
最大原子受力``` #!/bin/bash # eliminate forces of fixed atoms fix=$1 if test -z $1; then fix=0 fi # get force form OUTCAR awk -v fix="$fix" '/POSITION/,/drift/{ if($1~/^[0-9.]+$/&&$3>=fix) print $1,$2,$3,sqrt($4*$4+$5*$5+$6*$6i); else if($1=="total") print $0 }' OUTCAR >temp.f awk '{ if($1=="total") {print ++i,a;a=0} else {if(a<$4) a=$4} }' temp.f >force.conv #sed -i '1,9d' force.conv #rm temp.f tail -30 force.conv >temp.f #get dist from XDATCAR touch p1.conv p2.conv; touch dist.conv Num=`awk 'NR==7{for(i=1;i<=NF;i++) a=$i+a;}END{print a}' XDATCAR` Lnum=`wc XDATCAR|awk '{print $1}'` ((n=(Lnum-7)/(Num+1))) head -7 XDATCAR >p1.conv awk -v num="$Num" 'NR==9,NR==(num+1)+7{print $0}' XDATCAR >>p1.conv for((i=1;i<n;i++)) do head -7 XDATCAR >p2.conv ((n1=9+(Num+1)*i)) ((n2=(i+1)*(Num+1)+7)) #echo $n1 #echo $n2 sed -n "$n1,$n2"'p' XDATCAR >>p2.conv echo -e $i"\t"`dist.pl p1.conv p2.conv ` >>dist.conv done #plot gnuplot <<EOF set term dumb set xlabel 'Ion steps' set ylabel 'Dist (Angstrom)' plot 'dist.conv' w l t " Dist " set xlabel 'Ion steps' set ylabel 'Force (eV/Angstrom)' plot 'temp.f' w l t " Force " EOF rm force.conv dist.conv p1.conv p2.conv temp.f ```需要说明的是:因为脚本中涉及到很多距离计算,画图时间会略长。这里说所有原子所受最大力,其实不包括固定原子。vasp的力的收敛是忽略固定原子受力情况的。脚本中用到了dist.pl脚本。这里力只画了最后的30步,如需画更多步,更改脚本中的30即可。
注:
1.运行前要在Linux上安装dos2unix软件,运行dos2unix ××.sh将win的格式转为Linux的格式 2.conv_1.sh脚本运行需要系统中安装gnuplot作图软件,使用sudo apt install gnuplot安装即可 3.conv_2.sh脚本运行需要有VTST脚本库中的dist.pl脚本作为支持,下载该脚本并添加到环境变量中 #VTST# export PATH=$PATH:/home/host/VTST/vtstscripts-957 然后重新进入终端查看是否加载成功该脚本 (base) host@ubuntu:~/calculation/fang_calc/opt-1$ which dist.pl /home/host/VTST/vtstscripts-957/dist.pl 再运行该脚本即可 4.两个脚本运行命令均为bash ××.sh